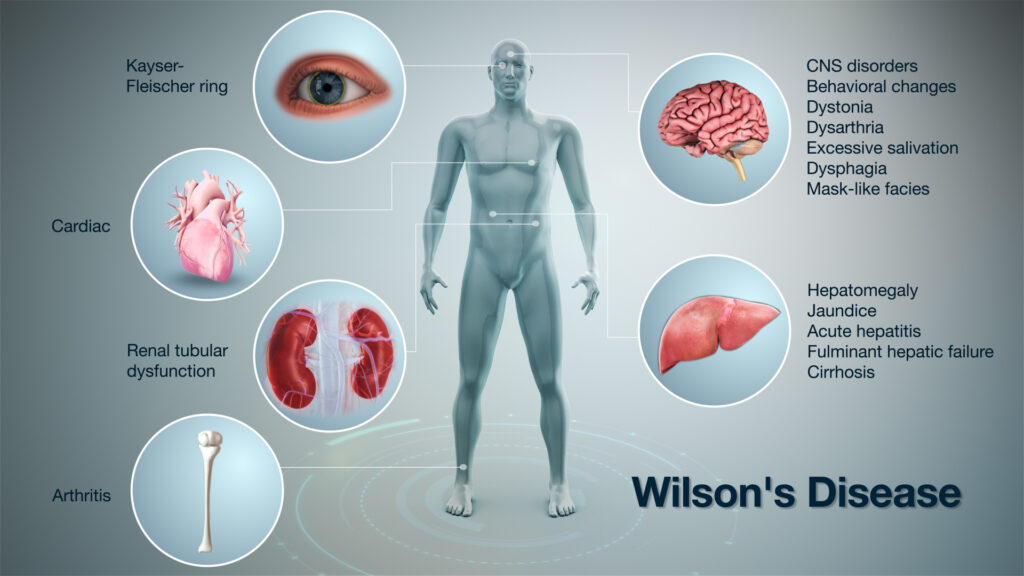
WHAT IS WILSON’S DISEASE?
Wilson’s disease, also known as hepatolenticular degeneration, is an uncommon inherited disorder that causes copper to accumulate in your liver, brain, and other important organs. People with Wilson’s disease are diagnosed between the ages of five years and thirty-five years, but it could also affect younger and senior citizens, as well.
Copper plays a crucial role in the development of strong or healthy nerves, bones, collagen, and skin pigment known as melanin. Generally, copper is absorbed from your food, and additional is excreted through a substance produced in your liver (bile).
But in people with Wilson’s disease, copper is not removed properly and instead builds up, possibly to a very dangerous stage. When detected early, Wilson’s disease is treatable, and several people with the disease live regular lives.
WILSON’S DISEASE SYMPTOMS
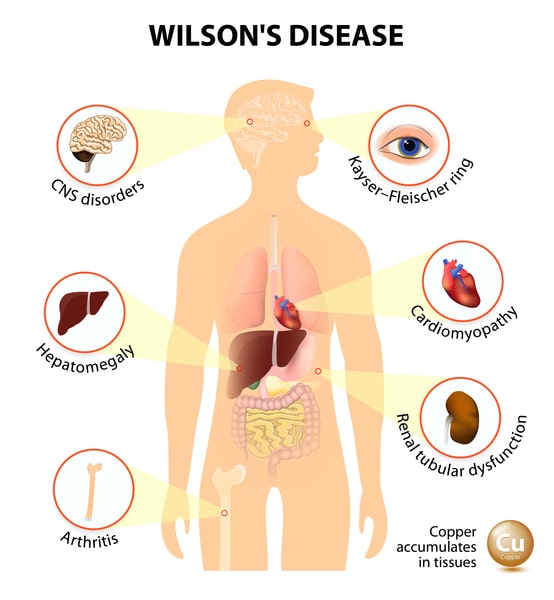
Wilson’s disease is present at the time of birth, but signs and symptoms do not appear till the copper accumulates in the brain, liver, or another organ. Signs and symptoms vary depending on the parts of your body damaged by the disease. They could include:
- Fatigue, lack of appetite, or abdominal pain
- Yellowing of the skin and the whiteness of the eyes (jaundice)
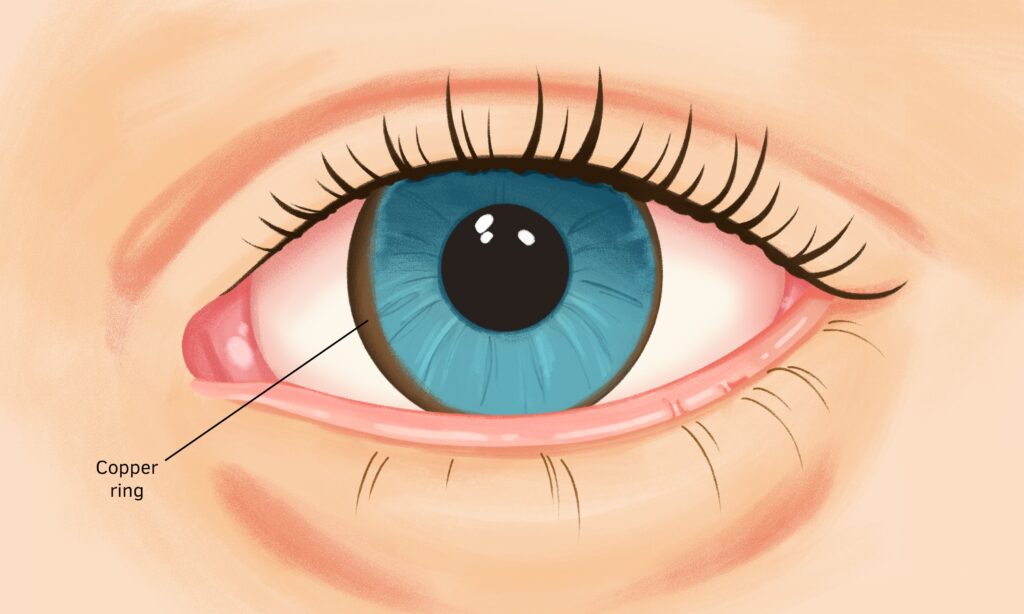
- Golden-brown eye discoloration (Kayser-Fleischer rings)
- Fluid build-up in the legs or abdomen
- Issues with speech, swallowing, or physical coordination
- Uncontrolled movements or muscle stiffness
WHEN SHOULD YOU SEE A DOCTOR?
Make an appointment with your doctor or primary care physician if you have signs and symptoms that worry you, particularly if a family member has Wilson’s disease.
WILSON’S DISEASE CAUSES
Wilson’s disease is inherited as an autosomal recessive trait, which simply means that to develop the disease you should inherit one copy of the defective gene from each parent. If you receive only one abnormal gene, you would not become sick yourself, but you are a carrier and could pass the gene to your children.
WILSON’S DISEASE RISK FACTORS
You could be at higher risk of Wilson’s disease if your parents or siblings have the condition. Ask your doctor or primary care physician whether you should undergo genetic testing to find out if you have Wilson’s disease. Diagnosing the condition as early as possible dramatically raises the chances of successful treatment.
WILSON’S DISEASE COMPLICATIONS
Untreated, Wilson’s disease could be fatal. Severe complications include:
Scarring of the liver (cirrhosis) – As liver cells try to make repairs to damage done by additional copper, scar tissue forms in the liver, making it harder for the liver to function.
Liver failure – This could happen unexpectedly (acute liver failure), or it could develop gradually over years. A liver transplant may be a treatment option.
Persistent neurological problems – Tremors, involuntary muscle movements, clumsy gait, and speech difficulties generally improve with treatment for Wilson’s disease. Although, some people have persistent neurological difficulty despite treatment.
Kidney problems – Wilson’s disease could affect the kidneys, leading to problems like kidney stones and an abnormal number of amino acids excreted in the urine.
Psychological problems – These may include personality changes, depression, irritability, bipolar disorder, or psychosis.
Blood problems – These may include destruction of red blood cells (hemolysis) leading to anemia and jaundice.
WILSON’S DISEASE DIAGNOSIS
Diagnosing Wilson’s disease can be difficult because its signs and symptoms are usually hard to tell from those of other liver illnesses, like hepatitis. Also, symptoms can evolve over time. Behavioral changes that come on gradually can be particularly hard to link to Wilson’s.
Doctors or primary care physicians rely on a combination of symptoms and test results to make the diagnosis. Tests and procedures for diagnosing Wilson’s disease include the following:
- Blood and urine tests – Blood tests could track your liver function and check the level of a protein that binds up copper in the blood (ceruloplasmin) and the level of copper in your blood. Your doctor or primary care physician also may want to measure the amount of copper excreted in your urine at the time of a twenty-four-hour period.
- Eye examination – Using a microscope with a high-intensity light source (slit lamp), an ophthalmologist checks your eyes for Kayser-Fleischer rings, which are caused by additional copper in the eyes. Wilson’s disease also is related to a type of cataract, known as a sunflower cataract that could be seen on an eye examination.
- Removing a sample of liver tissue for testing (biopsy) – Your doctor or primary care physician inserts a slim syringe through your skin, into your liver, and draws a small sample of tissue. A lab tests the tissue for excess copper.
- Genetic testing – A blood test could identify the genetic mutations that cause Wilson’s disease. Knowing the mutations in your family enables doctors or primary care physicians to screen siblings and start treatment before symptoms arise.
WILSON’S DISEASE TREATMENT
Your doctor or primary care physician may recommend medications known as chelating agents, which bind copper and then prompt your organs to release the copper into your bloodstream. The copper is then filtered through your kidneys and released into your urine.
Treatment then concentrates on preventing copper from building up again. For severe liver damage, a liver transplant may be required.
Wilson’s disease Medications
If you take medications for Wilson’s disease, treatment is for life. Medications include:
- Penicillamine (Cuprimine, Depen) – A chelating agent, penicillamine could cause severe side effects, including skin and kidney issues, bone marrow suppression, and deteriorating neurological symptoms. Penicillamine must be used cautiously if you have a penicillin allergy. It also keeps vitamin B-6 (pyridoxine) from working, so you will need to take a supplement in small doses.
- Trientine (Syprine) – Trientine works much like penicillamine but tends to cause few side effects. Still, neurological symptoms could worsen when taking trientine.
- Zinc acetate (Galzin) – This medication stops your body from absorbing copper from the food you eat. It is generally used as maintenance therapy to stop copper from building up again after treatment with penicillamine or trientine. Zinc acetate may be used as the main therapy if you cannot take penicillamine or trientine. Zinc acetate could cause stomach upset.
Your doctor or primary care physician may also recommend other medications for symptom relief.
Wilson’s disease Surgery
If your liver damage is serious, you may require a liver transplant. At the time of a liver transplant, a surgeon removes your diseased liver and replaces it with a healthy liver from a donor.
Most transplanted livers come from donors who are deceased. But in some cases, a liver could come from a living donor, like a family member. In that case, the surgeon removes your diseased liver and replaces it with a part of the donor’s liver.
If you or anyone you know is suffering from Wilson’s disease, our expert providers at Specialty Care Clinics will take care of your health and help you recover.
Call (469) 545-9983 to book a telehealth appointment for an at-home check-up.